Even for cells that are, in appearance, functionally homo-geneous, for instance neurons sharing shape, neurotrans-mitter, and spatial location, it is not proven thatthese resemblances correlate at the transcriptome level.
Concomitantly, very specific molecular markers are also scarce(see Okaty et al. for review). Cell mixtures are of limitedpower for analyzing the dynamics of gene expression, as onecannot distinguish between a change of the same amplitude inall the cells, a change taking place in a subset of the cells, or achange in the composition of the cell population.
Parallel analyses of single cells can resolve these questions,and change our view of cell populations. For example, show-ing phenotypic variations between individuals even whenthey are strongly related, such as cells growing together incell culture . These single-cell analyses are based mainlyon transcript imaging , PCR , or transcriptome analysis bymicroarray, and more recently, sequencing .
Some of these pioneering works have established thatinitiation of transcription can occur as bursts, for instancein Chinese hamster ovary cells , Dictyostelium ,Saccharomyces cerevisiae, etc. During these bursts,multiple copies of a transcript are generated in a short intervalfollowed by a period of rest that can be long when comparedwith the RNA’s half-life. Thus, in a population of cells where agiven protein is expressed, some cells may actually contain nocorresponding transcript. Combined with other sources of variation like the cell cycle, these biological mechanismscreate a large collection of different expression profiles, which other sources ofvariation like the cell cycle, these biological mechanismscreate a large collection of different expression profiles, whichnevertheless belong to the same cell type.
Collectively, the single-cell whole-transcriptome profilescontain information that can increase our understanding of the regulatory gene network in these cells (see Munsky et al. for review). In addition, they provide an opportunity for a data-driven identification and definition of cell types,especially given the increasing throughputs in processing andanalysis. This will add a new dimension to tissue sampleanalysis, allowing their description as a population of cellscharacterized by their cell types, similar to blood counts. Weterm these studies population transcriptomics. This approachhas direct applications in neuroscience, where there is cur-rently no comprehensive definition of cell types at the mol-ecular level , or in cancer research, where tumors areheterogeneous and involve cancer cells as well as somaticcells that have a stable genome but are induced into patho-logical functions by the tumor cells. In stem cell research, itwill allow a better understanding of the population dynamicswithin cell colonies or during trans-differentiation, where therole or effect of cell heterogeneity is still unclear.
Microfluidics is the study and the utilization of liquid flowsin small volumes, and this miniature environment allows thereduction of reagent volume, avoids loss of sample or samplecontamination, and provides a high throughput system forthe integration of multiple functions in a MicroTotal AnalysisSystem (mTAS) . Most microfluidics devices can be describedas tiny assembly lines where reactions are carried out as thesamples circulate from one specialized compartment to anotherthrough thin channels. These devices are embedded in carvedplatforms called chips, which are usually engineered by com-puter-aided design and produced using a system of photoli-thography, masks, and moulds [12] inspired by the methodsof production of Micro Electro Mechanical Systems (MEMS).Chip designs can be classified according to the method ofcreating isolated compartments, with either solid borders mate-rialized by microchambers, microwells, microvalves, etc., orfluid borders, in particular, microemulsions [13]. Whileeach cell comprises a compartment in itself, the methods fordetermining mRNA expression levels require cell lysis and,therefore, rely on the microfluidics device to isolate one cellper compartment.
In this review we will describe how single cell transcrip-tome analysis using microfluidic formats opens the doors tounderstanding cell population structures. First, we will focuson microemulsions (Fig. 2), which promise a high throughputsingle cell analysis. Next, we will review the all integrated’devices for single cell analysis of transcript expression levels.Finally, we will introduce new ways to investigate single celltranscriptomes and discuss the impact of high-throughputmicrofluidics.
Microdroplets are tiny liquidcompartments
Droplet microfluidics refers to the formation and transportof liquid nano- to microdroplets in a non-miscible carriermedium such as oil. Water-in-oil emulsions provide a popularand scalable system to produce these picoreactors of verysmall volume. Because they are generated at high frequency(several kHz) , they offer opportunities for high throughputand single cell analysis. The oil (mineral, silicone, or fluoro-carbon) that separates each droplet is non-miscible and inert.Surfactants prevent droplets from coalescing. Recently,fluorocarbon oils have gained popularity, thanks to the devel-opment of non-ionic, biocompatible surfactants.Microdroplets can be formed by focusing oil and water flows(Fig. 2 ). Single cell encapsulation is not traumatic forcells and allows cell survey, cell screening, multicellularorganism growth , or transfection .
Cells in suspension in the aqueous phase are encapsulatedstochastically following a Poisson law . As a consequence,a large number of empty droplets are produced under con-ditions that prevent the encapsulation of two cells in the samedroplet. To counter this drawback, different strategies havebeen adopted: inlet microchannel geometry design for self-organization of cells , on-demand laser driven drops,or improvement in droplet sorting. In the latter, after singlecell encapsulation, microdroplets are sorted by shear-inducedflow, fluorescence-activated dielectrophoresis (DEP),or piezoelectric effect .
Compartmentalization can also be achieved by simplespatial separation. For instance, Lin et al. created arraysof droplets covered by oil and separated with hydrophobicareas on a cover slip, requiring no special instruments. Moresophisticated strategies also exist. Using a commercially avail-able microdispersing instrument similar to inkjet printers,Liberski et al. printed an array of droplets of medium,in which they injected cells by printing a smaller volume inthe larger droplet medium. The droplets were covered by oil toprevent evaporation, but others achieved setups with no oil bymaintaining a locally high hygrometry . Lastly, otherwater-in-oil confinements have been investigated, for instancewith the chemistrode , where a succession of oil phases andaqueous ‘‘plugs’’ provides a temporal resolution, or the SlipChip , where channels materialized by the superposi-tion of two carved plates can be reversibly converted intonanoliter wells.
How to manipulate microdroplets?
Once the droplets are formed, it is difficult to introduce newreagents and carry out complex protocols. It is thereforenecessary in some cases to break up the emulsions. To main-tain the monoclonality of the reaction products after removingthe oil phase, that is, to keep the single-cell resolution afteropening the compartments, researchers have investigated twopossibilities. In the first, the reaction products are bound tobeads. The most prominent example is the 454 sequencingplatform . Alternatively, reaction products or cells havebeen encapsulated in microgels, such as agarose or poly-ethyleneglycol, gelated by polymerization with hyper-branched polyglycerol . After the reaction, the gel dropssolidify, thus trapping the amplicons. To avoid diffusionof PCR products out of the clonal agarose beads, Lenget al. conjugated the forward PCR primers directly withthe agarose.
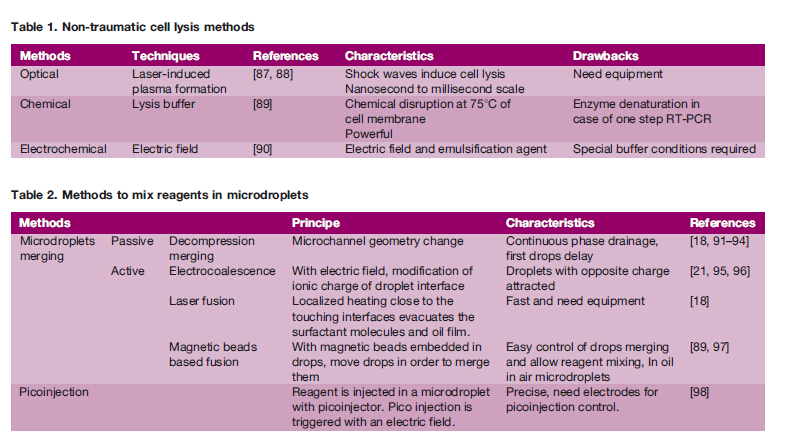
Given the stochasticity in gene expression levels, it isimportant that no artificial noise be added to the biologicalvariations, which may be informative. Therefore, the singlecell devices must avoid inducing stress pathways that arelikely to cause the transcription of some genes or the degra-dation of some transcripts. In addition to radical solutions likepre-fixation, the microfluidic designs can perform cell lysisquickly after tissue dissociation while maintaining highthroughput compared to hand picking. Table 1 summarizes some methods to snapshot the cellular state while preventingcells from expressing stress genes.
Multi-step reactions can be facilitated by delivery of newreagents for the next reaction steps. In microdroplet formats,reagents can be added by picoinjection or by merging droplets(Table 2), as demonstrated in the high-throughput single cellscreening reported by Brouzes et al. or in the microfluidicsystems for digital DNA amplification and counting usingrolling circle as shown by Mazutis et al. Droplet fusioncan be utilized not only for delivering contents; it can also beused for simple merging of droplets, for instance by electro-coalescence . Recently developed mesh-integrated arraysfor merging and storage offer the possibility to isolatesingle cell drops in picoliter compartments and to mixreagents for single cell-based assays. Incubation devices forcells or reactions are detailed in Table 3.
Altogether, these microdroplets are a toolbox in which theusual molecular biology reactions performed with microtubesand micropipettes are being reimplemented at a much smallerscale for high-throughput single-cell analysis pipelines. Butsometimes this approach, which causes some cells to bewasted, is not optimal. Proof-of-principle encapsulation devicesoften assume an unlimited input of cells, which is not realisticexcept in the cases of abundant primary cells or cultured cells.These experimental systems take the best from high throughputdesigns, but more focused designs are necessary when the cellsof interest are determined in advance or are very limited inquantity, as in the case of early embryos, and therefore shouldnot be lost. Devices using capture chambers instead of dropletsstill offer nanoliter-scale reactions, while reaching capture effi-ciencies as high as 5% . More complex combinations oftechnologies, with droplet sorting in the chips and cell pre-labeling (for instance by the injection or endocytosis ofmarkers) might not be available in the short-term.
Integrated microfluidics devices are likeminiaturized assembly lines
The construction of integrated devices for transcriptomeanalysis is challenging, as it requires the combination of thecritical steps previously described (Fig. 3). Zhong et al.used microchambers in which the mixing of reagents is easy,thus making the transition from RT to PCR very straightfor-ward. They obtained a RNA to cDNA conversion yield of 54% inthe device, compared to 12% with the same protocol executedin conventional tubes. Bontoux et al. designed a twocircular chamber device for HPRT and GAPDH RT-PCR ampli-fication from a single cell. But attempts at whole-transcriptome.
RNA amplification by template-switching failed, most likelydue to the lack of a purification step between RT and PCR; inthe absence of purification, the template-switching oligonu-cleotides compete with the universal PCR primers, thus inhib-iting the amplification reaction. Toriello et al. assembledthe most complete device, which includes cell selection in ananoreactor, capture, lysis, reverse-transcription, PCR, puri-fication of products, and separation.
By adjusting or producing components to fit their needs,multiple teams have assembled droplet-based devices ofincreasing complexity. It is expected that transcriptomeanalysis is also likely to be achieved using this compartmen-talization strategy. Multiple groups have already demonstratedRNA capture followed by cDNA synthesis and PCRamplification on the microdroplets format .
Transcriptome analysis includes the study of non-codingRNAs such as micro-RNAs, and White et al. estimatedmiRNA and mRNA molecule counts by qPCR in K562 cellsusing a multichamber design where the cells were captured,washed, and lysed, and cDNAs synthesized and then amplifiedby PCR with gene-specific primers. In addition to easy reagentflow control, microchambers also allow parallelization of reac-tions, and can process up to 300 cells per chip.
While already a multi-step protocol, PCR itself is a buildingblock for more complex workflows. A high-throughput singlecopy genetic amplification (SCGA) process has been developedby Kumaresan et al. In this process, cells or target DNAare encapsulated in nanoliter droplets containing functional-ized beads . The PCR products are bound to the beads, andanalyzed in flow cytometers. With a throughput of one milliondroplets per hour as a design goal, Zeng et al. reported theconception of Microfabricated Emulsion Generator Arrays(MEGA), in which single cells are encapsulated in dropletscontaining beads coated with forward PCR primers, and fluo-rescently labeled reverse PCR primers. After cell lysis and PCR,the beads are bound with fluorescently labeled amplicon if thePCR was positive. The result of the experiment is then read bycounting in a flow cytometer, and the beads exhibiting fluor-escent labels are chosen for each target gene.
Multiplexing of devices is achieved in a straightforward man-ner by replicating a building block on the lab chip. Thisstrategy fits particularly well in cases where the reagents orthe starting materials are pre-arranged in standard formatssuch as the 96-well plate. But alternatives are needed whenaiming at higher orders of magnitude. By preparing a reagentdroplet library in which each droplet contains a specific primerpair and these are fused to droplets containing single-cells,Tewhey et al. developed an enrichment device for large-scale targeted sequencing. Incubation is conducted outsidethe device in a thermocycler and the products are recovered bybreaking the emulsion with a destabilizer solution. Anothermodular strategy, with two devices connected by an incu-bation channel functioning as a delay line , was describedby Agresti et al.This featured cell encapsulation, dropincubation, and sorting for directed evolution. In anotherscreen, Brouzes et al. fused a library of single-chemicaldroplets to droplets containing single human monocytic U937cells. The chemical contents of the merged droplets weretracked by optical coding with a combination of fluoro-chromes, a strategy that might be employed later in a differentcontext, for instance to record treatments, spatial origin ortemporal series.
Devices that can prepare large numbers of single cells for geneexpression analyses open the way to a new representation ofthe cell’s transcriptome. With current studies of cell mixtures,transcript expression levels are measured relative to eachother using methods such as quantitative PCR, microarrays,cDNA sequencing (RNA-Seq, etc.) or in situ hybridization.
Consequently, our tools and concepts are centered on theseapproaches in which a significant fold-change will be apositive result. However, relative expression levels are anincomplete readout of a simpler parameter: the number oftranscripts present in a sample. Single-cell whole-transcriptome counting technologies aim atabsolute precision. One key challenge is to prevent loss oftemplates. Losses seriously complicate the interpretation ofthe data since the absence of signal would not exclude that thecells were expressing the RNA. Two strategies to avoid theselosses differ in their trade-off between complexity and sensi-tivity. Single-molecule sequencing provides direct countsbut offers no protection against losses. The aim of thisapproach is, therefore, to simplify the protocol as much aspossible, for instance by sequencing RNA by synthesis insteadof using a cDNA intermediate . On the other hand, inmethods in which the transcripts are amplified, usually ascDNAs, passive adsorption and similar losses will not cause atranscript to become undetectable. However, expression levelsmay be biased by the amplification unless the cDNAs thatoriginate from the same mRNA molecule can be recognizedand considered as a single count. This has been done byKivioja et al. with the introduction of unique moleculeidentifiers (UMIs). Alternatively, the use of random primers inthe reverse transcription will produce a collection of cDNAswhere the random priming site itself can be considered as aunique molecular identifier, such as in CAGEscan , pro-vided that there is no strand displacement. This strategy couldbe generalized to any semi-anchored transcriptome analysistechnology, for instance when the anchor is the RNA 30 -end,via oligo-dT priming or linker ligation.
qPCR and imaging contribute independentobservations
Before reaching a critical mass and becoming well established,sequence-based data will need to be confirmed by othermethods. In particular, it is still unclear how many RNAmolecules are expected in single cells. Beyond the effects ofstochastic expression, surprisingly large variations can befound in subpopulations of cells that would otherwise beconsidered of the same type. Molecule counting has beenapproximated by quantitative PCR using a titration curve.
For instance, Sta˚hlberg et al. analyzed single astrocytesand single neurosphere cells by qPCR (50 cycles with opti-mized primers) and found two sub-populations distinguishedby their low or high expression of marker genes, where thehighest counts exceeded 40,000 for the Vimentin or GFAPtranscripts. Taken together, these two subpopulations couldexplain more complex expression patterns as the sum of twosimpler lognormal-style distributions. Molecule counting hasalso been implemented by another application of microflui-dics, digital PCR . In this droplet-based embodiment,up to one million reactions are parallelized, leading to anaverage error of 15%, to be compared with qPCR, whichproduces a quantitative error of 100% in just a single cycle(as it doubles the product once per cycle).
Sequencing methods and qPCR have common limitations.In particular, they are not in situ and are, therefore, unable todistinguish nascent RNAs from mature RNAs unless pulse-labeling or run-on methods are implemented. This wouldbe challenging in microfluidics devices. Second, they areinherently limited by the incompleteness of the reverse-tran-scription step . Therefore, transcript counting by imaging is an important complementary technique, providing a tech-nologically independent line of evidence to the sequencingand qPCR-based measurements. This technology is beingactively developed and is progressing in throughput .
Imaging-based analysis of mRNA expression levels is ame-nable to microfluidics miniaturization. By confining singlecells in channels and tracking them across cell divisions,Rowat et al. studied the diversity of modes of expressionin yeast. Using three genes as models and GFP fusions as areporter system, they could demonstrate constitutive (Rps8b),inherited (Pho84), and heterogeneous (Hsp12) expressionmodes. This diversity has been reported earlier and is oftenmodeled as bursts of transcription. However, many of thepioneering studies were focused on a small number of genes.Using a device for high throughput imaging by batches of 96,Taniguchi et al.covered the whole genome and reportedno correlation between protein and RNA levels in Escherichiacoli. Importantly, Taniguchi et al. derived a model whereGamma distribution parameters were interpreted as transcrip-tion rate and protein burst size. This lack of correlation wasalso observed by Raj et al. who noted that while the RNApolymerase II gene was expressed in bursts, the bursts in othergenes were not synchronous, suggesting that the transcriptionnoise is buffered at the protein level. Indeed, using a desta-bilized GFP, Raj et al. observed correlation between proteinand mRNA levels, which could not be observed with the usual,more stable GFP. Other mechanisms also decouple the expres-sion levels of proteins and mRNAs. For instance, mRNA can bestored in P-bodies and stress granules directly after transcrip-tion .
Methods for highly multiplexed transcript counting byimaging have also been developed. For instance,NanoStrings were used with single cells by Khan et al.to demonstrate that the olfactory enhancers function toincrease the number of cells expressing transcripts, asopposed to the levels of transcript expression in each cell.This work also showed that hybridization-based technologiescan reach a high stringency, since the authors coulddistinguish 577 different olfactory receptors despite stronghomologous sequences.to demonstrate that the olfactory enhancers function toincrease the number of cells expressing transcripts, asopposed to the levels of transcript expression in each cell.This work also showed that hybridization-based technologiescan reach a high stringency, since the authors coulddistinguish 577 different olfactory receptors despite stronghomologous sequences.
Cells that look similar can contain differentamounts of transcripts
While studying the distribution of transcript expression levels,it will be essential to ensure that technical noise is not addedto the biological variations. In particular, a high confidencelevel is needed to distinguish between the true absence of atranscript and the failure of its detection. Indeed, Bengtssonet al. showed that RT-PCR noise is stronger than biologicalvariations for transcripts present in less than 100 mRNA or 20cDNA copies. They also reported striking differences inreverse-transcription efficiencies, ranging between 2% and99%. Miniaturization has the potential to mitigate this prob-lem. For instance, in the device of Zhong et al. the minimaldetectable number of molecules was four, compared to 17 in abulk assay. More recently, Reiter et al. showed that inlymphocytes, the noise associated with reverse-transcriptionwas far greater than the technical noise caused by the presence of cell debris in the mixture, which suggests thatreaction volumes could be reduced in micro-compartments.Altogether, the methods based on reverse-transcription willneed to be carefully calibrated by comparing them with themeasurements based on imaging.
Zenklusen et al. used 50 probes to study transcriptioninitiation in S. cerevisiae cells. Nascent transcripts are detectedas a nuclear signal, of which the intensity is a multiple of thesingle spots in the cytoplasm. They estimated $60,000mRNAs per cell, which is 3–6 times higher than previousestimates. In this study of three house-keeping genes, lessthan 10% of the cells did not contain the target RNA, and thedata had a Poisson distribution. Study of the MDN1 geneindicated pauses longer than one minute between transcrip-tion initiations. The data fits models of bursts but it is import-ant to note that other models were also compatible. The studyof other genes suggests that not all genes are transcribed inbursts. For instance, POL1, regulated by cell cycle, did notshow bursts as most cells did not contain more than onenascent RNA, in contrast to the SAGA þ TATA-controlled genePDR5.
Unlike relative expression levels, expression counts aredramatically modified at each cell division, where they wouldbe halved if the division were symmetric. However, with theexception of very tightly controlled molecules such as chro-matids, most molecules are not equally segregated betweendaughter cells. Huh and Paulsson have demonstrated thatmodels of random partition noise can also predict stochasticexpression counts, that are often solely modeled as the out-come of transcription bursts in the literature. Moreover, theyalso show that molecular mechanisms such as aggregation orsub-cellular localization in vesicles can further increase thatnoise. In fast-dividing cells, partition noise of low-expressedupstream regulators might also explain the stochasticity ofexpression. In addition to dilution by cell division, levels ofRNAs are also decreased by degradation. This can also bestudied by imaging, for instance by comparing the outputof 50 and 30 probes .
Heterogeneity within a population can also be caused byother mechanisms, such as ageing. Taken on more than ageneration, all cell divisions are potentially asymmetric, asone daughter cell may inherit a half where the molecules orthe organelles have been synthesized at an earlier generationand therefore had more time to accumulate damage. In linewith this hypothesis, Wang et al. showed in E. coli thatcells do not die by exponential decay, but rather, the cells thatinherit the halves that have been passed down over manygenerations are distinguishable from others by their higherprobability to die. Altogether, cell division has direct implica-tions on the heterogeneity of the transcriptome.
Charles Plessy Ã, Linda Desbois, Teruo Fujii and Piero Carninci



.jpg)
.jpg)

.jpg)